Revisión actualizada de la patogénesis, los fenotipos clínicos, los métodos diagnósticos y las estrategias terapéuticas en las enfermedades ampollares autoinmunes, con énfasis en los avances recientes
Lugones Editorial©
Las enfermedades bullosas autoinmunes (autoimmune bullous diseases, AIBD), también conocidas como enfermedades ampollares autoinmunes, constituyen un grupo de trastornos caracterizados por la formación de ampollas en la piel y las mucosas como consecuencia de autoanticuerpos dirigidos contra proteínas estructurales de la epidermis o de la unión dermoepidérmica. Estos autoanticuerpos alteran la integridad epitelial y provocan la formación de ampollas y erosiones.
A pesar de los avances en la comprensión de su patogénesis y tratamiento, las enfermedades ampollares autoinmunes continúan planteando desafíos diagnósticos y terapéuticos debido a su complejidad inmunopatológica y a la heterogeneidad de sus manifestaciones clínicas.
En esta revisión, los autores sintetizan el conocimiento actual sobre la patogénesis de las enfermedades ampollares, sus fenotipos clínicos, los métodos diagnósticos y las estrategias terapéuticas, con especial énfasis en los avances recientes y en las oportunidades traslacionales.
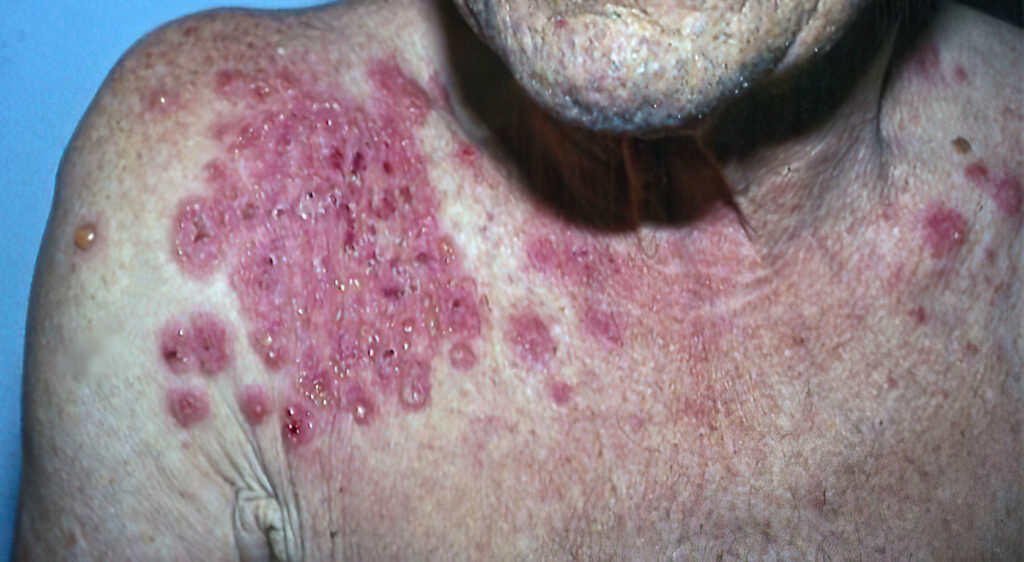
Las enfermedades ampollares autoinmunes continúan planteando desafíos diagnósticos y terapéuticos debido a su complejidad inmunopatológica y a la heterogeneidad de sus manifestaciones clínicas
Introducción
Las enfermedades bullosas autoinmunes (autoimmune bullous diseases, AIBD), también denominadas enfermedades ampollares autoinmunes, se clasifican de manera general en dos grandes grupos:
- Trastornos del pénfigo (por ejemplo, pénfigo vulgar y pénfigo foliáceo), caracterizados por ampollas intraepidérmicas producidas por autoanticuerpos dirigidos contra proteínas desmosómicas.
- Trastornos penfigoides (por ejemplo, penfigoide ampollar y penfigoide de membranas mucosas), caracterizados por ampollas subepidérmicas debidas a una respuesta autoinmune dirigida contra componentes de la zona de la membrana basal.
El diagnóstico de las AIBD requiere un enfoque multimodal que integre la evaluación clínica, la histopatología, la microscopía de inmunofluorescencia y la detección serológica de autoanticuerpos. La inmunofluorescencia directa (DIF) continúa siendo el estándar de oro diagnóstico, mientras que los ensayos serológicos, como el ensayo inmunoabsorbente ligado a enzimas (ELISA), permiten confirmar la presencia de autoanticuerpos y monitorizar la actividad de la enfermedad.
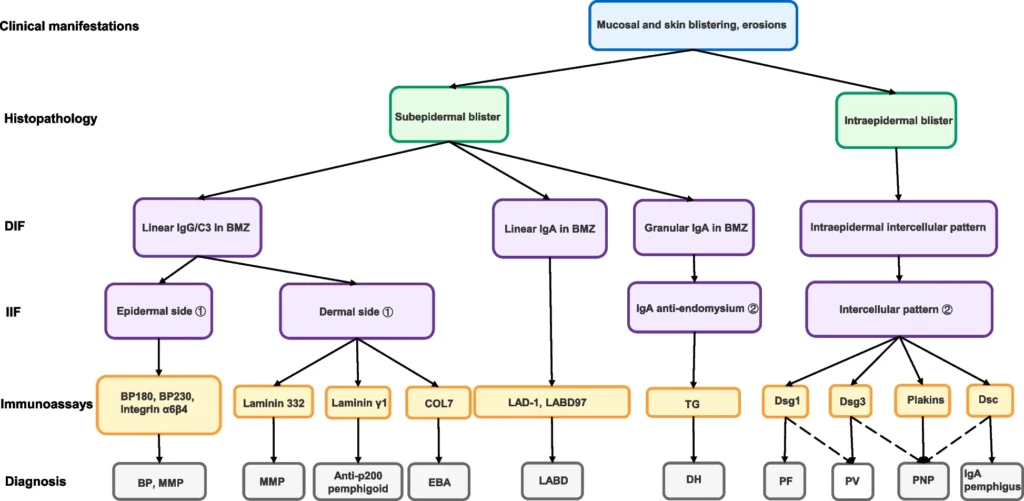
Diagrama de flujo de diagnóstico para enfermedades bullosas autoinmunes
Pénfigo
El pénfigo es un grupo de enfermedades autoinmunes ampollosas que afectan la piel y las mucosas. Se caracterizan por la producción de autoanticuerpos contra proteínas desmosómicas, principalmente desmogleína 1 (Dsg1) y desmogleína 3 (Dsg3), lo que provoca pérdida de adhesión entre queratinocitos (acantólisis) y formación de ampollas. Los subtipos más importantes son pénfigo vulgar (PV) y pénfigo foliáceo
- Epidemiología
El pénfigo vulgar es el subtipo más frecuente y representa aproximadamente el 70% de los casos. Su incidencia es de alrededor de 2,8 casos por millón de personas al año. El pénfigo foliáceo es menos común y puede presentarse en forma esporádica o endémica, con una prevalencia aproximada de 10 casos por millón.
- Factores genéticos y ambientales
La susceptibilidad al pénfigo está influida por factores genéticos, especialmente alelos específicos del complejo HLA, que varían según la población. Además, se han detectado autoanticuerpos en niveles bajos en familiares de primer grado de pacientes afectados, lo que sugiere una predisposición genética.
Entre los factores ambientales asociados se encuentran ciertos medicamentos, infecciones bacterianas y virales, estrés, y algunos factores dietéticos, especialmente alimentos ricos en compuestos tiol. En el pénfigo foliáceo endémico también influyen factores como exposición a insectos, trabajo al aire libre y condiciones de vida específicas.
- Patogénesis
El pénfigo es una enfermedad mediada por linfocitos T y B autorreactivos. Las células B, activadas por células T CD4+, se diferencian en células plasmáticas que producen autoanticuerpos contra Dsg1 y/o Dsg3. Estos anticuerpos se unen a los desmosomas de los queratinocitos, alteran la adhesión celular y provocan acantólisis y formación de ampollas.
En la respuesta inmunitaria se observa un predominio de citocinas Th2, participación de células Th17 en fases activas y una reducción de células T reguladoras, lo que favorece la pérdida de tolerancia inmunológica. La hipótesis de “múltiples golpes” propone que varios autoanticuerpos actúan conjuntamente para producir la alteración de la adhesión celular.
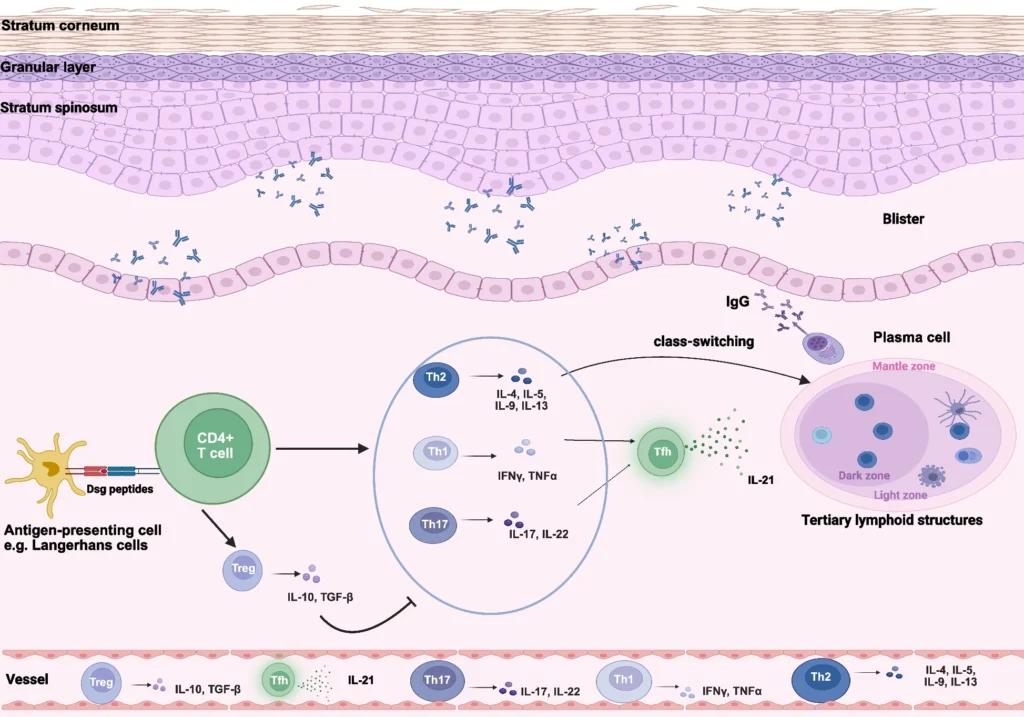
Patogénesis del pénfigo vulgar
- Características clínicas
Aunque el PV y el PF comparten mecanismos inmunológicos similares, difieren en sus antígenos diana y en su presentación clínica.
. En el pénfigo vulgar, los autoanticuerpos se dirigen principalmente contra Dsg3 y a veces Dsg1. La enfermedad suele comenzar con lesiones dolorosas en la mucosa oral, que pueden dificultar la alimentación. Posteriormente aparecen ampollas cutáneas flácidas que se rompen con facilidad, especialmente en zonas sometidas a fricción.
. En el pénfigo foliáceo, los autoanticuerpos se dirigen contra Dsg1, por lo que las lesiones se limitan a la epidermis superficial. Clínicamente se observan ampollas superficiales que se rompen rápidamente, formando erosiones con costras, generalmente en zonas seborreicas como la cara y el cuero cabelludo.
- Diagnóstico
El diagnóstico se basa en la correlación clínica, histológica e inmunológica. La biopsia cutánea muestra acantólisis intraepidérmica: suprabasal en el PV y en la capa granular en el PF. La inmunofluorescencia directa demuestra depósitos de IgG y/o C3 con patrón reticular entre queratinocitos. Además, la detección sérica de anticuerpos anti-Dsg1 y anti-Dsg3 mediante ELISA ayuda a confirmar el diagnóstico y a monitorizar la actividad de la enfermedad.
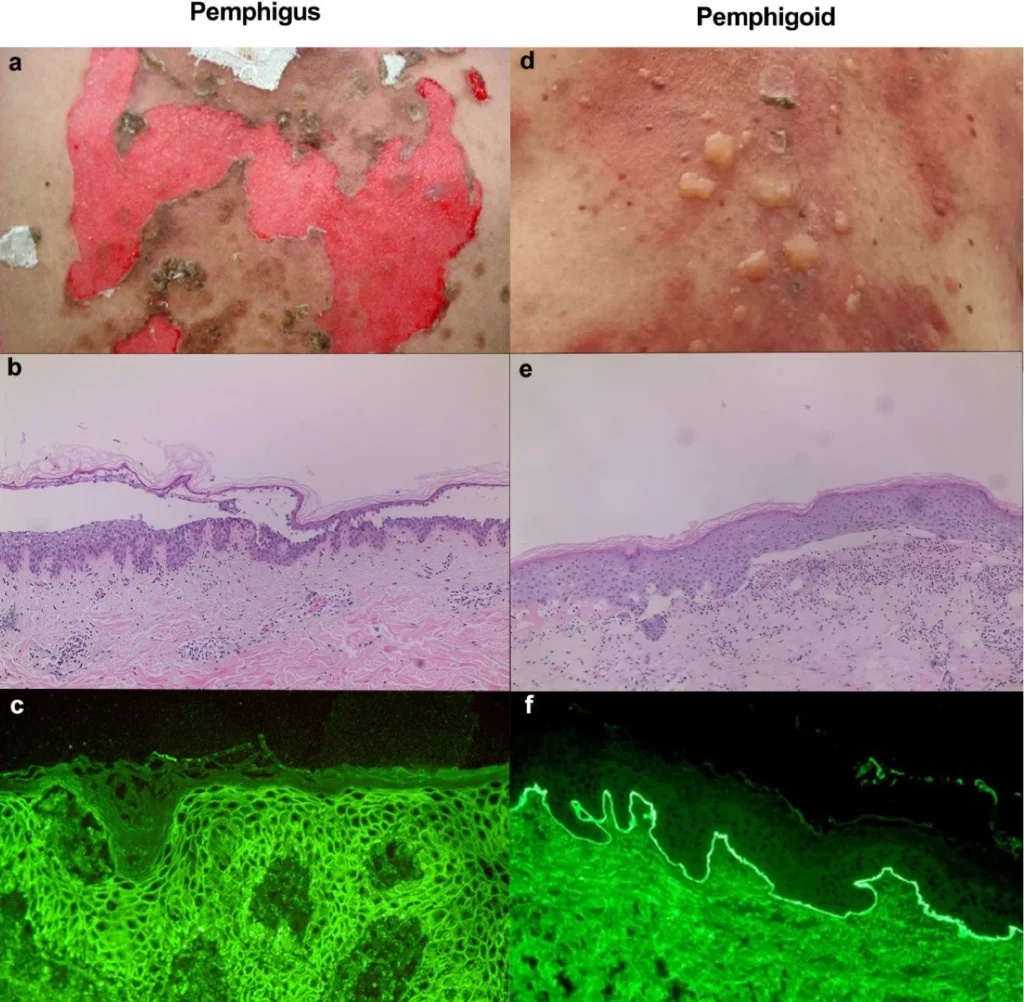
Principales hallazgos clínicos y de laboratorio en pénfigo y penfigoide
- Tratamiento tradicional
El tratamiento se basa principalmente en corticosteroides sistémicos, que siguen siendo la terapia inicial para la mayoría de los pacientes. En casos leves se emplean dosis moderadas, mientras que en enfermedad moderada o grave se requieren dosis más altas. Una vez controlada la enfermedad, la dosis se reduce gradualmente para evitar recaídas.
Frecuentemente se utilizan inmunosupresores adyuvantes, como micofenolato de mofetilo o azatioprina, para mejorar el control de la enfermedad y permitir reducir la dosis de corticosteroides. En casos graves o resistentes pueden emplearse terapias adicionales como inmunoglobulina intravenosa, plasmaféresis o inmunoadsorción.
- Terapias dirigidas
Las terapias dirigidas buscan actuar sobre mecanismos inmunológicos específicos y reducir los efectos adversos de los tratamientos tradicionales. El rituximab, un anticuerpo monoclonal anti-CD20 que elimina linfocitos B, ha demostrado una alta eficacia y actualmente se considera terapia de primera línea en el pénfigo moderado a grave.
Otras terapias en investigación incluyen otros anticuerpos anti-CD20, inhibidores de BTK, inhibidores de BAFF, bloqueo del receptor FcRn, inhibidores de JAK y terapias celulares dirigidas contra células B autoreactivas.
- Manejo multidisciplinario
El pénfigo es una enfermedad crónica que requiere seguimiento prolongado y manejo multidisciplinario. Los dermatólogos evalúan la actividad de la enfermedad y monitorizan los niveles de autoanticuerpos, mientras que otros especialistas supervisan las complicaciones sistémicas y los efectos secundarios de la inmunosupresión.
Las infecciones, especialmente neumonía y sepsis, constituyen una de las principales causas de mortalidad, por lo que la vigilancia clínica y la prevención de infecciones son fundamentales. El apoyo psicológico también es importante debido al impacto que tiene la enfermedad crónica en la calidad de vida de los pacientes.
Pénfigo paraneoplásico
El pénfigo paraneoplásico (PNP) es una enfermedad ampollosa autoinmune rara asociada a neoplasias, caracterizada por lesiones mucocutáneas graves y afectación multiorgánica. Inicialmente se consideró un subtipo de pénfigo, pero actualmente se reconoce como una entidad distinta debido a su patogénesis compleja, manifestaciones sistémicas y fuerte relación con tumores. En 2001 se propuso el término síndrome autoinmune multiorgánico paraneoplásico (PAMS) para reflejar mejor su naturaleza sistémica.
- Epidemiología
El PNP es extremadamente raro, con una incidencia estimada de menos de un caso por millón de habitantes al año. Existe cierta predisposición genética relacionada con alelos específicos del HLA, aunque la evidencia es limitada debido al bajo número de casos. La enfermedad está estrechamente asociada a neoplasias, especialmente tumores hematológicos o linfoproliferativos, como la enfermedad de Castleman o la leucemia linfocítica crónica.
- Patogénesis
La patogénesis del PNP se basa en la producción de autoanticuerpos generados en el contexto de la neoplasia, que reaccionan de forma cruzada con proteínas epiteliales. Estos autoanticuerpos se dirigen contra múltiples antígenos, incluyendo proteínas desmosómicas y hemidesmosómicas, como desmoplacina, envoplacina, periplacina, BP230 y desmogleínas 1 y 3.
Además de la inmunidad humoral, la inmunidad mediada por células desempeña un papel importante, con infiltración de linfocitos T citotóxicos y liberación de citocinas proinflamatorias como interferón-γ y TNF-α, que contribuyen al daño epitelial.
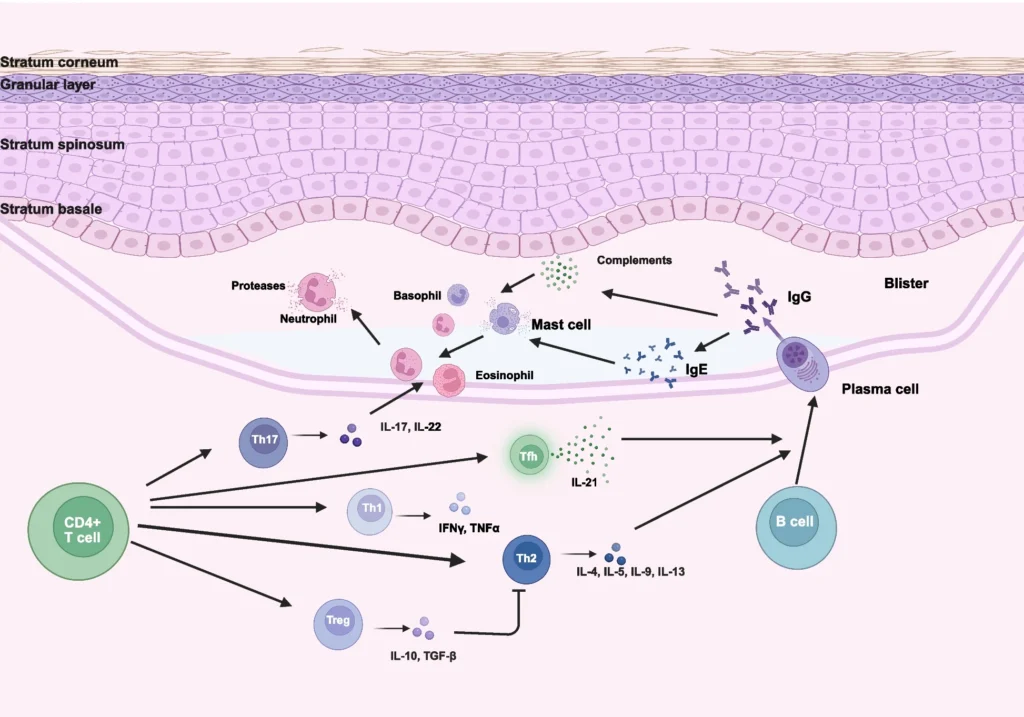
Patogénesis del penfigoide bulloso
- Características clínicas
El PNP se caracteriza por mucositis erosiva grave y persistente, junto con lesiones cutáneas polimórficas, que pueden incluir ampollas, dermatitis liquenoide o lesiones similares al eritema multiforme. A diferencia del pénfigo clásico, la enfermedad suele asociarse a manifestaciones sistémicas, siendo especialmente relevante la bronquiolitis obliterante, una complicación pulmonar potencialmente mortal.
Histológicamente puede observarse acantólisis, necrosis de queratinocitos y dermatitis de interfaz liquenoide. La inmunofluorescencia directa suele mostrar depósitos de IgG y C3 con patrón intercelular en la epidermis o lineal en la zona de la membrana basal. Las pruebas serológicas permiten detectar autoanticuerpos contra envoplacina, periplacina u otras proteínas relacionadas.
- Tratamiento
El tratamiento se centra principalmente en identificar y tratar la neoplasia subyacente, cuya resección o control puede mejorar el curso de la enfermedad. Los corticosteroides sistémicos constituyen la base del tratamiento farmacológico, aunque su eficacia puede ser limitada en la mucositis o en la afectación pulmonar.
Con frecuencia se emplean inmunosupresores adyuvantes, como azatioprina, metotrexato o ciclosporina, así como inmunoglobulina intravenosa en casos resistentes.
- Terapias dirigidas
Las terapias dirigidas se utilizan especialmente en PNP asociado a neoplasias de células B. El rituximab, anticuerpo monoclonal anti-CD20, puede reducir la producción de autoanticuerpos y mejorar la enfermedad. En casos refractarios se han utilizado otras terapias inmunológicas, como alemtuzumab o anticuerpos dirigidos contra células T.
La bronquiolitis obliterante es una complicación grave que puede requerir tratamientos inmunomoduladores adicionales y, en casos avanzados, trasplante pulmonar.
- Manejo multidisciplinario
Debido a su carácter sistémico, el PNP requiere un enfoque multidisciplinario. Dermatólogos, hematólogos u oncólogos, neumólogos y otros especialistas deben colaborar para tratar la enfermedad cutánea, la neoplasia asociada y las complicaciones sistémicas.
El uso de inmunosupresores aumenta el riesgo de infecciones oportunistas, por lo que es esencial un seguimiento clínico estrecho. La coordinación entre diferentes especialidades es fundamental para mejorar el pronóstico de esta enfermedad compleja.
Enfermedad penfigoidea
Las enfermedades penfigoides constituyen un grupo de trastornos ampollares autoinmunes subepidérmicos caracterizados por autoanticuerpos dirigidos contra proteínas estructurales de la zona de la membrana basal (BMZ), esenciales para la adhesión dermoepidérmica. Entre los principales antígenos implicados se encuentran BP180 (colágeno XVII), BP230, laminina 332, integrina α6β4 y colágeno tipo VII.
Los subtipos más relevantes incluyen el penfigoide bulloso (PB), el penfigoide de membranas mucosas (PMM), la epidermólisis bullosa adquirida (EBA) y el penfigoide anti-p200, cada uno con manifestaciones clínicas y características inmunológicas particulares.
- Epidemiología y factores predisponentes
El penfigoide bulloso es el subtipo más frecuente y afecta principalmente a adultos mayores, con una edad media de inicio entre los 66 y 83 años. Su incidencia aumenta con la edad y se estima entre 2,4 y 21,7 casos por millón de habitantes al año. El penfigoide de membranas mucosas es menos común y se caracteriza por un curso crónico con tendencia a cicatrización, mientras que la EBA y el penfigoide anti-p200 son entidades raras.
La susceptibilidad genética se ha asociado con determinados alelos HLA de clase II, y diversos factores ambientales pueden actuar como desencadenantes, incluyendo medicamentos, infecciones, traumatismos, radioterapia o exposición a radiación ultravioleta.
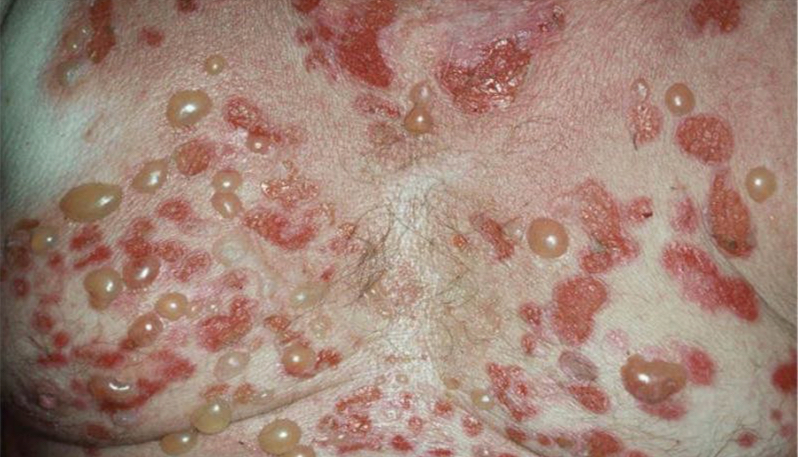
Las enfermedades penfigoides constituyen un grupo de trastornos ampollares autoinmunes subepidérmicos caracterizados por autoanticuerpos dirigidos contra proteínas estructurales de la zona de la membrana basal
- Patogénesis
Las enfermedades penfigoides se desarrollan por una respuesta autoinmune contra proteínas de la membrana basal, que conduce a la separación dermoepidérmica y la formación de ampollas. Los autoanticuerpos activan el sistema del complemento y desencadenan el reclutamiento de células inflamatorias, como neutrófilos y eosinófilos, que liberan enzimas proteolíticas capaces de degradar componentes estructurales de la unión dermoepidérmica.
Las citocinas asociadas a la respuesta Th2, especialmente IL-4, IL-13 e IL-31, contribuyen a la inflamación y al prurito característico.
- Características clínicas y diagnóstico
Las manifestaciones clínicas varían según el subtipo. El penfigoide bulloso se presenta típicamente con ampollas tensas sobre piel eritematosa o urticarial, que suelen afectar tronco y extremidades. El penfigoide de membranas mucosas compromete principalmente mucosas, en especial la cavidad oral y los ojos, y puede producir cicatrices. La EBA se caracteriza por fragilidad cutánea y ampollas en áreas expuestas al traumatismo, con tendencia a cicatrización.
Histológicamente se observan ampollas subepidérmicas, y la inmunofluorescencia directa demuestra depósitos lineales de IgG y/o C3 en la unión dermoepidérmica. Las pruebas serológicas permiten detectar autoanticuerpos específicos, como anti-BP180, anti-BP230 o anti-colágeno tipo VII.
- Tratamiento
Los corticosteroides constituyen la base del tratamiento, ya sea en forma tópica o sistémica según la gravedad. En casos refractarios o recurrentes pueden utilizarse inmunosupresores como azatioprina, metotrexato o micofenolato mofetilo.
En los últimos años se han incorporado terapias dirigidas, como rituximab, dupilumab u omalizumab, que actúan sobre diferentes vías inmunológicas implicadas en la enfermedad.
- Manejo multidisciplinario
Debido a su curso crónico y a la posible afectación de mucosas y otros órganos, el manejo de las enfermedades penfigoides requiere un enfoque multidisciplinario, con participación de dermatólogos y otros especialistas según la localización de las lesiones, para optimizar el control de la enfermedad y prevenir complicaciones.
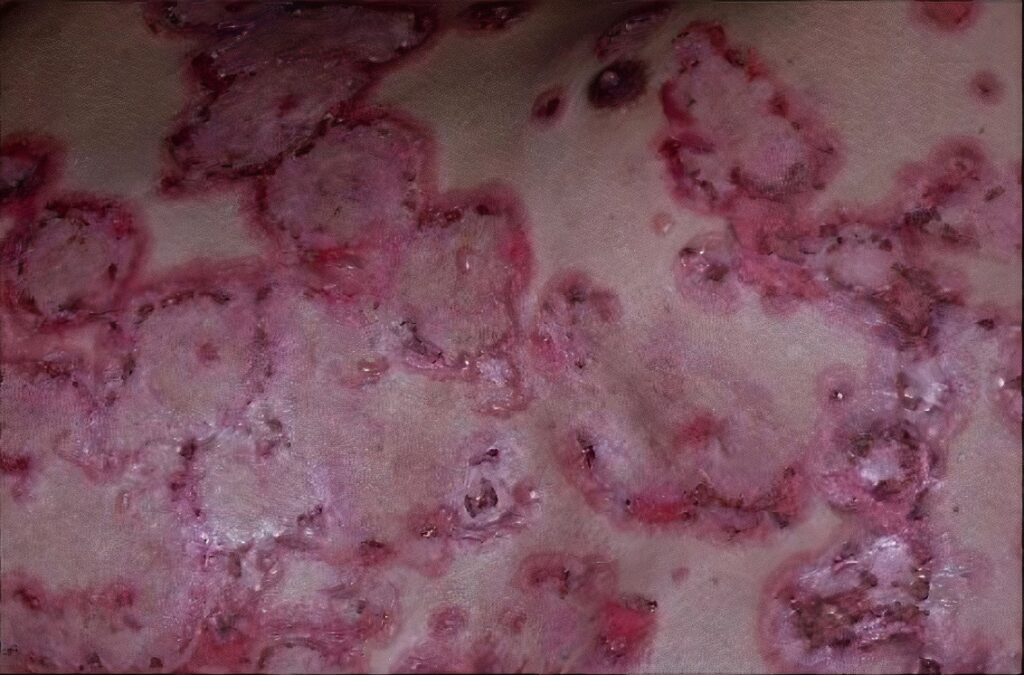
El penfigoide bulloso se presenta típicamente con ampollas tensas sobre piel eritematosa o urticarial, que suelen afectar tronco y extremidades
Enfermedades autoinmunes ampollares relacionadas con IgA
Las enfermedades ampollares autoinmunes relacionadas con IgA incluyen el pénfigo IgA, la dermatosis ampollar lineal por IgA (LABD) y la dermatitis herpetiforme (DH). Se caracterizan por la presencia de autoanticuerpos IgA dirigidos contra proteínas de adhesión cutánea, lo que conduce a inflamación y formación de ampollas. Aunque comparten el mecanismo mediado por IgA, difieren en sus antígenos diana, características clínicas y asociaciones sistémicas.
- Epidemiología y factores predisponentes
El pénfigo IgA es una enfermedad rara que puede aparecer a cualquier edad, aunque se observa con mayor frecuencia en adultos entre la cuarta y sexta década de vida. La LABD presenta una distribución bimodal, con un pico en la infancia temprana y otro en adultos mayores.
La dermatitis herpetiforme tiene una incidencia estimada de 0,4 a 3,5 casos por 100.000 habitantes al año y está estrechamente relacionada con la sensibilidad al gluten y con predisposición genética, especialmente con determinados alelos HLA implicados en el procesamiento de antígenos derivados de la gliadina.
- Patogénesis
El mecanismo patogénico central es la producción de autoanticuerpos IgA que desencadenan inflamación cutánea mediante la activación de células inmunitarias y el reclutamiento de neutrófilos.
En el pénfigo IgA, los autoanticuerpos suelen dirigirse contra desmocolina 1, lo que provoca alteraciones en la adhesión entre queratinocitos. En la LABD, los anticuerpos IgA se dirigen contra componentes de la zona de la membrana basal, particularmente fragmentos derivados de BP180.
La dermatitis herpetiforme se desarrolla como consecuencia de la respuesta inmunológica frente al gluten. La ingestión de gliadina induce la formación de autoanticuerpos IgA contra la transglutaminasa tisular, que posteriormente pueden reaccionar con la transglutaminasa epidérmica, generando depósitos cutáneos de IgA y desencadenando inflamación.
- Características clínicas y diagnóstico
Las manifestaciones clínicas varían según la enfermedad. El pénfigo IgA suele presentarse con vesículas o ampollas que evolucionan hacia pústulas, localizadas principalmente en tronco, cuero cabelludo y áreas intertriginosas.
La LABD se caracteriza por ampollas tensas o vesículas agrupadas, a menudo con una disposición característica en “cúmulo de joyas”, y puede afectar tanto piel como mucosas.
La dermatitis herpetiforme se manifiesta con pápulas, vesículas y erosiones intensamente pruriginosas, distribuidas de forma simétrica en superficies extensoras y nalgas.
El diagnóstico se basa en la histopatología y en la inmunofluorescencia directa, que muestra depósitos de IgA con patrones característicos: intercelular en el pénfigo IgA, lineal en LABD y granular en la dermatitis herpetiforme.
- Tratamiento
La dapsona constituye el tratamiento de primera línea para la mayoría de las enfermedades ampollares mediadas por IgA, debido a su efecto sobre la infiltración neutrofílica. Los corticosteroides sistémicos pueden utilizarse como terapia adyuvante, aunque su eficacia es variable.
En casos refractarios pueden emplearse otros fármacos inmunomoduladores, como sulfonamidas, colchicina, metotrexato o ciclosporina. En la dermatitis herpetiforme, además del tratamiento farmacológico, es fundamental mantener una dieta estricta sin gluten de por vida, lo que permite controlar la enfermedad y mejorar el pronóstico.
Terapias biológicas como rituximab o inhibidores del TNF-α han mostrado resultados prometedores en casos resistentes, aunque su uso aún se encuentra en evaluación.
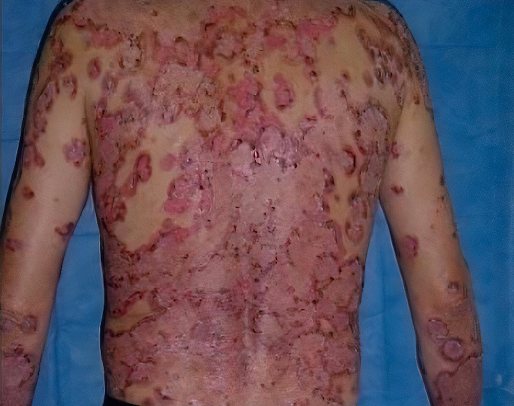
La dapsona constituye el tratamiento de primera línea para la mayoría de las enfermedades ampollares mediadas por IgA, debido a su efecto sobre la infiltración neutrofílica
Conclusiones
Las enfermedades ampollares autoinmunes (AIBD) constituyen un grupo heterogéneo de trastornos caracterizados por respuestas inmunitarias dirigidas contra proteínas clave de adhesión cutánea. En los últimos años se han logrado avances significativos en la comprensión de sus mecanismos patogénicos, particularmente en relación con el papel de los autoanticuerpos, las células inmunitarias y los antígenos diana implicados en los distintos subtipos.
Estos conocimientos han favorecido la evolución del tratamiento, pasando de estrategias inmunosupresoras convencionales hacia terapias dirigidas, que buscan modular de forma más específica las vías inmunológicas responsables de la enfermedad.
De cara al futuro, la integración de enfoques de biomedicina molecular y análisis multiómico podría permitir identificar firmas de autoanticuerpos específicas, comprender mejor el microambiente inmunológico de estas enfermedades y desarrollar terapias biológicas más precisas. Estos avances tienen el potencial de mejorar el diagnóstico, optimizar las estrategias terapéuticas y avanzar hacia una medicina más personalizada en el manejo de las AIBD.
Diagnóstico de enfermedades ampollares
Fuente
Feng X, Zheng H, Wang M, et al. Autoimmune bullous diseases: pathogenesis and clinical management. Mol Biomed. 2025 15;6:30.